
New therapies for inborn errors of metabolism
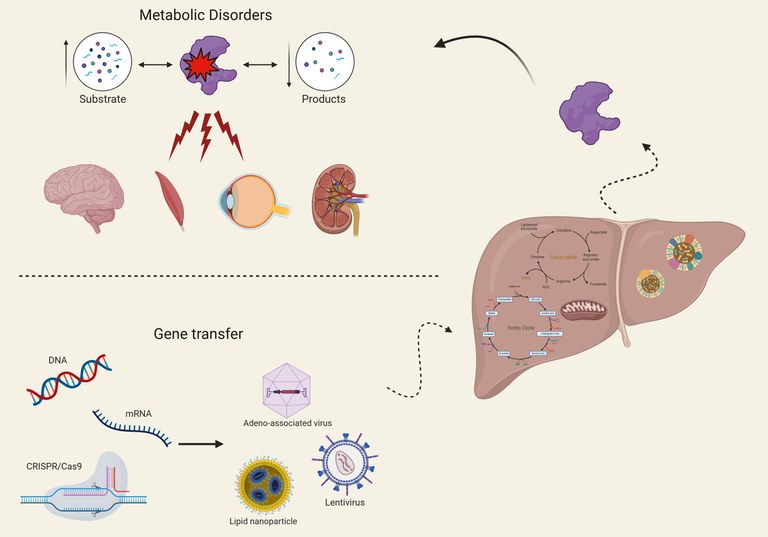
We are investigating the regulation of urea cycle enzymes and how this knowledge could impact the development of novel therapies for urea cycle disorders. We also use this knowledge to better understand the pathogenesis and for treatment of complex multifactorial diseases. Among inborn errors of metabolism, we are interested in organic acidemias, primary hyperoxalurias and glycogen storage disease besides the urea cycle disorders. We are investigating both gene transfer and small molecule drugs for these disorders.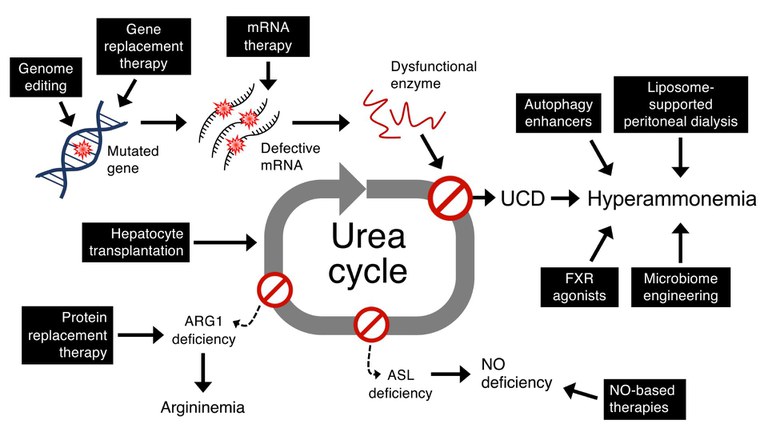
The ultimate goal of this research is to translate mechanistic discoveries into the clinical arena via clinical research performed at the Unit of Innovative Therapies for Genetic Diseases of the Federico II University Hospital of Naples. My clinical research includes an active program on liver-directed gene therapy with ongoing clinical trials.
Nicola Brunetti-Pierri is the Head of the TIGEM Translational Incubator, which aims to develop large-scale in vivo preclinical studies for the treatment of different inherited disorders by using standardized and optimized protocols to facilitate translation from the bench to the bedside.











Quote
As a paediatrician and geneticist, the overall goals of my research are to understand how defects in biochemical pathways result in disease and to develop novel therapeutic approaches for inborn errors of metabolism.
Additional Funding
-
Oxalosis & Hyperoxaluria Foundation (OHF) - Clinically relevant animal models of Primary Hyperoxaluria Type 3 (2024-2026)
- SGF - Social Good Fund - AAV-mediated liver-directed gene therapy for gyrate atrophy of the choroid and retina (2024-2026)
- AFM - Gene therapy for Wolman disease (2022-2023)